- Visibility 109 Views
- Downloads 42 Downloads
- DOI 10.18231/j.ijooo.2024.009
-
CrossMark

Bilateral episodic rise of intraocular pressure: A case of pigment dispersion syndrome
- Author Details:
-
 Arti Singh *
Arti Singh *
-
Upasana Singh
-
Srishti Nagarajan
Introduction
Pigment Dispersion Syndrome is an autosomal dominant disorder with onset in mid- 20s[1] characterized by disruption of iris pigment epithelium and deposition of pigment granules on the anterior segment structures. Some clinical signs that are consistently present in white patients such as iris transillumination defects and anterior iris stromal pigment dusting are, however, not as common in blacks. Although most patients experiencing an episode of pigment dispersion are asymptomatic, extreme photophobia, ocular pain, redness, and blurred vision may occur.
Case Presentation
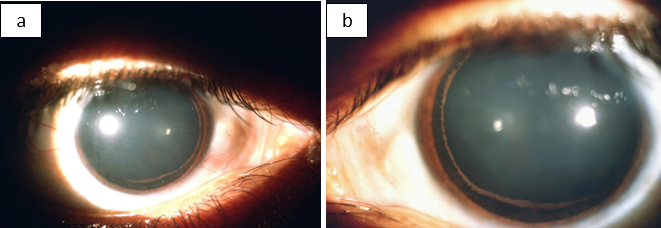
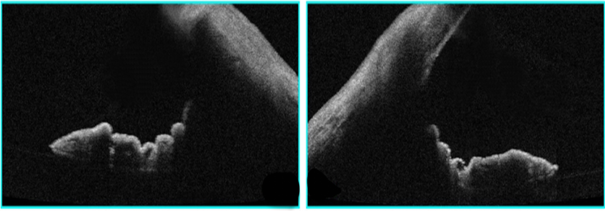
A 28-year-old male patient presented to our hospital with chief complaints of on and off episodes of coloured halos and blurring of vision in both eyes for 2 years. His best corrected visual acuity in each eye was 6/9 P. On slit lamp examination diffuse corneal oedema was present hence applanation tonometry done which measured an IOP of 50 mm of Hg in both eyes. Tablet acetazolamide was prescribed, and patient was followed on next day. Mild corneal oedema with 26 mm of Hg IOP was noted. Krukenberg’s spindle was seen in both eyes ([Figure 1]) and mid peripheral iris transillumination defect was absent on slit lamp observation. Examination of anterior chamber revealed increase depth, both in central and peripheral areas with no posterior synechiae, cells and flare. Upon dilatation, a Zentmayer’s ring or Scheie stripe ([Figure 2]a,b) was noted. Fundoscopy revealed cup disc ratio 0.8 and 0.7 in right and left eye respectively. Gonioscopy examination revealed concave configuration or posteriorly bowing of iris, hyperpigmented trabecular meshwork and Sampaolesi line in both eyes ([Figure 3]). ASOCT also revealed peripheral concave configuration of iris ([Figure 4]) with central corneal thickness below the normal range (463m and 471m in right and left eye respectively). A diagnosis of bilateral pigmentary glaucoma was entertained, and Bimatoprost (0.01%) eye drops once in a day in both eyes was started. One-week later, corneal oedema completely disappeared and IOP was 16 mmHg in both eye and he was advised to continue topical medication.


Discussion
Pigment Dispersion Syndrome is an autosomal dominant disorder with onset in mid- 20s [1] characterized by disruption of iris pigment epithelium. The released pigment granules are carried by aqueous convection currents and deposited on anterior segment structures including the corneal endothelium, iris surface, trabecular meshwork (TM), lens surfaces, and zonules.[2] Although most patients experiencing an episode of pigment dispersion are asymptomatic, extreme photophobia, ocular pain, redness, and blurred vision may occur.[3] PDS affects 2–4% of patients during the second to fourth decades of life. The higher prevalence of PDS in young adults makes this group more prone to have POHT.One study has thus reported that after 10 years of progression, the amount of pigment on the posterior surface of the cornea decreased, and in certain patients who had been treated for pigmentary glaucoma, the disease remained stable after discontinuation of treatment. Monitoring the IOP must be rigorous since each 1 mmHg rise above 21 mmHg increases the risk for developing PG by 1.4 times. [3] The typical position and the vicinity of the zonular bundles with the mid-peripheral iris and frequent friction with iris movements suggest the cause of loss of pigment from the posterior iris. By means of convective current of the aqueous, pigments get accumulated on the structures of the anterior part of the eye [4] like corneal endothelium, anterior capsule of lens, trabecular meshwork and pigment deposits on the posterior capsule along the insertion of the zonules are referred to as the Scheie stripe or Zentmayer line. [5] The diagnostic triad of PDS includes corneal pigmentation (Krukenberg Spindle), mid-peripheral iris transillumination defects and dense trabecular pigmentation. [6] It can lead to secondary open angle glaucoma called pigmentary glaucoma (PG) with estimated risk between 35% to 50%.[7] Male gender, black race, severe myopia, and Krükenberg spindles were identified as possible risk factors. It usually affects men at an earlier age and they had a higher proportion in the glaucoma group; and they required more aggressive glaucoma therapy.[8] The loss of trabecular cells, fusion of trabecular lamellae with collapse of intertrabecular spaces, increase in extracellular material and obliteration of canal in various amounts around the circumference of eyes can be the cause of rise in intraocular pressure in PDS.[9] The clinical course of PG is divided into 3 phases [10] i.e. active pigment dispersion, conversion to glaucoma and regression phase. High resolution anterior segment anterior segment ultrasound biomicroscopy can show mid peripheral iris concavity, irido-zonular contact or irido-ciliary process contact in untreated eyes of PDS. [11] Persons with pigment dispersion syndrome also have an abnormally high incidence of lattice degeneration of retina and retinal detachment.[12] Treatment is typically medical which includes prostaglandin analogues, alpha adrenergic agents and beta blockers. Laser trabeculoplasty is more effective in PG. [13] Laser peripheral iridotomy (PI) has been proposed as a treatment option for PDS but there is no scientific evidence as of yet to associate PI as a treatment for PDS. [14]
Conclusion
PDS affects 2–4% of patients during the second to fourth decades of life. The higher prevalence of PDS in young adults makes this group more prone to have POHT. Monitoring of IOP therefore becomes an integral part in every follow up.
As the medical treatment proves to be beneficial enough, laser trabeculoplasty can also be sought as an alternative treatment option in Pigmentary Glaucoma.
Source of Funding
None.
Conflict of Interest
None.
References
- DG Campbell, RM Schertzer. Pathophysiology of pigment dispersion syndrome and pigmentary glau- coma. Curr Opin Ophthalmol 1995. [Google Scholar]
- DK Roberts, MA Chaglasian, RE Meetz. . Optom Vis Sci 1997. [Google Scholar]
- Y Siddiqui, RDT Hulzen, JD Cameron, DO Hodge, DH Johnson. . Am J Ophthalmol 2003. [Google Scholar]
- WE Evans, RE Odom, EJ Weenas. Krukengerg’s spindle: a study of 202 collected cases. Arch Ophthalmol 1941. [Google Scholar]
- N Niyadurupula, DC Broadway. Pigment dispersion syndrome and pigmentary glaucoma--a major review. Clin Exp Ophthalmol 2008. [Google Scholar]
- G Scuderi, M T Contestabile, L Scuderi, A Librando, V Fenicia, S Rahimi. Pigment dispersion syndrome and pigmentary glaucoma: a review and update. Int Ophthalmol 2018. [Google Scholar]
- N Niyadurupula, DC Broadway. Pigment dispersion syndrome and pigmentary glaucoma-a major review. Clin Exp Ophthalmol 2008. [Google Scholar]
- SM Farrar, MB Shields, KN Miller, CM Stoup. Risk factors for the development and severity of glaucoma in the pigment dispersion syndrome. Am J Ophthalmol 1989. [Google Scholar]
- R Ritch. A unification hypothesis of pigment dispersion syndrome. Trans Am Ophthalmol Soc 1996. [Google Scholar]
- SA Gandolfi, N Ungaro, P Mora, C Sangermani, TM Shaarawy, MB Sherwood, RA Hitchings, JG Crowston. Pigmentary glaucoma. Glaucoma: medical Diagnosis and Therapy 2009. [Google Scholar]
- SD Potash, C Tello, J Liebmann, R Ritch. Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology 1994. [Google Scholar]
- J Gottanka, DH Johnson, F Grehn, E Lütjen-Drecoll. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma 2006. [Google Scholar]
- MK Kuchle, NX Nguyen, CY Mardin, GO Naumann. Effect of neodinium : YAG laser iridotomy on number of aqueous melanin granules in primary pigment dispersion syndrome. Graefes Arch Clin Exp Ophthalmol . 2001. [Google Scholar]
- J Buffault, B Leray, A Bouillot, C Baudouin, A Labbé. Role of laser peripheral iridotomy in pigmentary glaucoma and pigment dispersion syndrome: A review of the literature. J Fr Ophtalmol 2017. [Google Scholar]